Data Production
Once we receive new sequencing, the samples are processed into CRAMs/gVCFs and stored until ready to joint genotype call into a project level VCF (pVCF). GCAD will generate one pVCF containing all new and previously generated gVCFs once per year. These pVCFs then undergo ADSP QC and are deposited into NIAGADS for the research community for access. The tables below provide an update on what data has been processed.
| Dataset Round | Number of Samples | WGS/WES | Status | CRAM/gVCF Release Date | Preview pVCF Release Date | QC pVCF Release Date |
|---|---|---|---|---|---|---|
| Round 1 | 4,789 | WGS | Complete | July 2018 | NA | October 2018 |
| Round 2 | 20,504 | WES | Complete | February 2020 | NA | September 2020 |
| Round 3 | 16,905 | WGS | Complete | March 2021 | March 2021 | October 2021 |
| Round 4 | 36,361 | WGS | Complete | October 2022 | October 2022 | August 2023 |
| Round 5 | 58,507 | WGS | In Process | November 2024 | November 2024 | Anticipate late 2025 |
| Round 6 | ~76,000 | WGS | In Process | Anticipate mid 2026 | Anticipate mid 2026 | TBD |
| Release | Project Name | WGS/WES | Total Samples | Data Received | gVCFs Generated |
|---|---|---|---|---|---|
| R2 | ADSP-DiscoveryCC-WES | WES | 10939 | Oct-17 | Jun-19 |
| R2 | WHICAP-AA-WES | WES | 1131 | Apr-18 | Jun-19 |
| R2 | ADGC-AA-WES | WES | 3225 | Jul-17 | Jun-19 |
| R2 | WHICAP-Hispanic-WES | WES | 1829 | Jun-18 | Jun-19 |
| R2 | WHICAP-Caucasian-WES | WES | 958 | Jul-18 | Jun-19 |
| R2 | MIAMI-WES | WES | 114 | Oct-18 | Jun-19 |
| R2 | CBD-WES | WES | 361 | May-20 | Jun-19 |
| R2 | PSP-WES | WES | 704 | Apr-20 | Jun-19 |
| R2 | Knight ADRC-WES | WES | 661 | Mar-18 | Jun-19 |
| R2 | ZORAN-WES | WES | 77 | Oct-18 | Jun-19 |
| R2 | FASe-WES | WES | 1104 | Nov-18 | Jun-19 |
| R1;R3 | ADSP-ExtensionFam-WGS | WGS | 444 | Dec-16 | Nov-19 |
| R1;R3 | ADSP-DiscoveryFam-WGS | WGS | 584 | Jan-14 | Nov-19 |
| R1;R3 | ADSP-ExtensionCC-WGS | WGS | 2959 | Jun-16 | Jan-20 |
| R1;R3 | ADNI-WGS1 | WGS | 809 | Sep-12 | Nov-19 |
| R3 | CurePSP-Macrogen-WGS | WGS | 886 | Jan-18 | Oct-19 |
| R3 | UPitt-Batch1-WGS | WGS | 209 | Jul-17 | Oct-19 |
| R3 | CurePSP-UCLA-WGS | WGS | 408 | Feb-18 | Nov-19 |
| R3 | APOE-Extreme-WGS | WGS | 885 | Oct-18 | Dec-19 |
| R3 | FUS-ADCAutopsy-WGS | WGS | 2772 | Sep-19 | Oct-19 |
| R3 | CacheCounty-WGS | WGS | 207 | Jul-17 | Dec-19 |
| R3 | Knight ADRC-WGS | WGS | 77 | Nov-18 | Jan-20 |
| R3 | FASe-WGS | WGS | 91 | Nov-18 | Jan-20 |
| R3 | Genentech-NACC-WGS | WGS | 137 | Jan-19 | Oct-19 |
| R3 | CurePSP-USUHS-WGS | WGS | 617 | Jul-19 | Aug-19 |
| R3 | FUS-PR1066-WGS | WGS | 1517 | Sep-19 | Oct-19 |
| R3 | FUS-ADGCAA-WGS1 | WGS | 1923 | Sep-19 | Oct-19 |
| R3 | FUS-ADNI-WGS2 | WGS | 757 | Sep-19 | Nov-19 |
| R3 | FUS-HIHG-Brainbank-WGS | WGS | 92 | Sep-19 | Feb-20 |
| R3 | AMPAD-ROSMAP-WGS | WGS | 730 | Jan-19 | Feb-20 |
| R3 | AMPAD-ADMSSM-WGS | WGS | 344 | Feb-19 | Nov-19 |
| R3 | AMPAD-ADMAYO-WGS | WGS | 252 | Feb-19 | Feb-20 |
| R3 | FUS-Stanford-WGS | WGS | 214 | Nov-19 | Feb-20 |
| R4 | FUS-EOAD-WGS | WGS | 1009 | Apr-20 | Jun-20 |
| R4 | FUS-MHAS-WGS | WGS | 2653 | Apr-20 | Jul-20 |
| R4 | FUS-Ibadan-WGS | WGS | 965 | Jun-20 | Aug-20 |
| R4 | FUS-CWAutopsy-WGS | WGS | 176 | Aug-20 | Dec-21 |
| R4 | FUS- ADC-AA-WGS | WGS | 306 | Aug-20 | Sep-20 |
| R4 | FUS- ADC- Amerindian-WGS | WGS | 84 | Aug-20 | Sep-20 |
| R4 | FUS-EFIGA-WGS | WGS | 1086 | Aug-20 | Nov-20 |
| R4 | FUS- Miami-BrainBank-WGS | WGS | 315 | Sep-20 | Dec-20 |
| R4 | LASI-DAD-WGS | WGS | 2767 | Nov-20 to Oct-21 | Nov-21 |
| R4 | FUS-PRADI-WGS | WGS | 744 | Jan-21 | Feb-21 |
| R4 | FUS-CUADI-WGS | WGS | 100 | Jan-21 | Feb-21 |
| R4 | FUS-AMISH-WGS | WGS | 1055 | Jan-21, May-21 | Seq-21 |
| R4 | FUS-StepAD2-WGS | WGS | 128 | Mar-21 | July-21 |
| R4 | FUS-REAAADI-WGS | WGS | 740 | Mar-21 | Sep-21 |
| R4 | FUS-Rapid Decline-WGS | WGS | 171 | Mar-21 | Aug-21 |
| R4 | EOAD-WGS | WGS | 3176 | May-21 | June-21 |
| R4 | TARCC-WGS | WGS | 1018 | Apr-21 | July-21 |
| R4 | FUS-AD Peru-WGS | WGS | 252 | May-21 | July-21 |
| R4 | FUS-WRAP | WGS | 1434 | Oct-21 | Nov-21 |
| R4 | FUS-ADC Hispanics | WGS | 1214 | Sep-21 | Nov-21 |
| R4 | FUS-NOMAS | WGS | 778 | Sep-21 | Nov-21 |
| R5 | UPitt-Kamboh2-WGS | WGS | 209 | Sep-19 | Oct-19 |
| R5 | ASPREE WGS | WGS | 2795 | Jan-21 | May-24 |
| R5 | GARD-WGS | WGS | 2007 | Aug-21 | Oct-23 |
| R5 | EOAD2 | WGS | 1264 | Nov-21 | Jan-22 |
| R5 | FUS-A4 | WGS | 3385 | Feb-22 | May-23 |
| R5 | FUS-RAPID-DECLINE2-WGS | WGS | 65 | Mar-22 | May-23 |
| R5 | FUS-CNSA-WGS | WGS | 274 | Mar-22 | May-23 |
| R5 | Wellderly-WGS | WGS | 1207 | May-22 | Mar-23 |
| R5 | FUS-ADNI-WGS-3 | WGS | 622 | Jun-22 | Feb-23 |
| R5 | FUS-ADC-AA-WGS-2 | WGS | 767 | Jun-22 | Feb-23 |
| R5 | FUS-PeADI2-WGS | WGS | 265 | Aug-22, Jan-24 | Apr-23, Feb-24 |
| R5 | FUS-CuADI2-WGS | WGS | 27 | Aug-22 | May-23 |
| R5 | FUS-NOMAS2-WGS | WGS | 30 | Aug-22 | May-23 |
| R5 | FUS-MexicanAPP-PSEN-WGS | WGS | 66 | Aug-22 | Jul-23 |
| R5 | FUS-KBASE-WGS | WGS | 603 | Sep-22 | Feb-23 |
| R5 | Apoe-longitudinal | WGS | 88 | Jan-23 | Jan-23 |
| R5 | FUS-STEPAD3-WGS | WGS | 125 | Jan-23 | Mar-23 |
| R5 | AmyloidImaging-WU | WGS | 1113 | Feb-23, Nov-23 | Aug-23, Dec-23 |
| R5 | AmyloidImaging-Pitt | WGS | 1195 | Feb-23 | Nov-23 |
| R5 | FUS-Amish2-WGS | WGS | 96 | Apr-23 | May-23 |
| R5 | UAB-ADRC-WGS | WGS | 17 | May-23 | Jul-23 |
| R5 | FUS-CWAutopsy2-WGS | WGS | 40 | May-23 | Jul-23 |
| R5 | FUS-ASPREE-WGS | WGS | 342 | May-23 | Oct-23 |
| R5 | FUS-EFIGA2-WGS | WGS | 1787 | Jun-23,Feb-24 | Nov-23, May-24 |
| R5 | FASe2-WGS | WGS | 659 | Nov-23 | Dec-23 |
| R5 | APOE-Extremes2-WGS | WGS | 27 | Nov-23 | Mar-24 |
| R5 | HABS-HD-WGS | WGS | 1362 | Jan-24 | Mar-24 |
| R5 | CU-Hispanics | WGS | 2632 | Dec-23 | Jun-24 |
| R6 | FUS-REGARDS-WGS | WGS | 2161 | Feb-22 | Oct-23 |
| R6 | Portuguese WGS | WGS | 374 | Aug-23 | Mar-24 |
| R6 | FUS-IDIBAPS-WGS | WGS | 826 | Sep-23 | Nov-23 |
| R6 | FUS-ISAVRAD-WGS | WGS | 1130 | Apr-24, Sep-24, Oct-25 | Oct-24, Feb-26 |
| R6 | FUS-WHICAP-WGS | WGS | 414 | Feb-24 | Jan-25 |
| R6 | FUS-PeADI3-WGS | WGS | 295 | Dec-24, Sep-25 | Jan-25, Oct-25 |
| R6 | HABS-HD2-WGS | WGS | 2941 | Sep-24, May-25 | Apr-25, TBD |
| R6 | FUS-READD-HI-WGS | WGS | 2361 | Nov-24, Sep-25, Apr-26 | May-25, Jan-26, Jul-26 |
| R6 | AZAPOE2-WGS | WGS | 240 | Feb-25, Apr-26 | May-25, Apr-26 |
| R6 | GARD2-WGS | WGS | 1976 | May-25 | Jul-25 |
| R6 | FUS-Rush-WGS | WGS | 1551 | Jun-25 | Oct-25 |
| R6 | FUS-CUADI3-WGS | WGS | 456 | Aug-25, Nov-25, Apr-26 | Oct-25, Jan-26, Aug-26 |
| R6 | RUSH-Brazil-WGS | WGS | 5523 | Jul-25, Feb-26, Jul-26 | Feb-26, Apr-26, Jul-26 |
| R6 | FUS-GAPP-WGS | WGS | 625 | Jun-25 | Mar-26 |
| R6 | FUS-KBASE2-WGS | WGS | 490 | Jun-25, Apr-26 | Mar-26, Jul-26 |
| R6 | FUS-LOS-WGS | WGS | 99 | Jun-25 | Nov-25 |
| R6 | FUS-ACAD-WGS | WGS | 521 | Aug-25 | Oct-25 |
| R6 | FUS-MHAS2-WGS | WGS | 2084 | Jan-26 | TBD |
| R6 | FUS-GLASS-WGS | WGS | 186 | Apr-26 | Jun-26 |
| R7 | FUS-READD-AA-WGS | WGS | 1871 | Dec-24, Apr-25, Sep-25, Apr-26 | Feb-25, Apr-25, Mar-26, TBD |
| R7 | FUS-READD-AF-WGS | WGS | 1451 | Apr-25, Sep-25, Apr-26 | May-25, Dec-25, TBD |
| R7 | MLSFH-WGS | WGS | 3431 | Mar-26 | TBD |
| R7 | MENA-WGS | WGS | 641 | Apr-26 | TBD |
| R7 | FUS-VALIANT-WGS | WGS | 228 | Apr-26 | TBD |
| R7 | FUS-MexicanAPP-PSEN2-WGS | WGS | 295 | May-26 | TBD |
Pipeline
All pipelines are co-developed with ADSP investigators.
VCPA pipeline
The SNP/Indel Variant Calling Pipeline and data management tool (VCPA) is the official pipeline used for processing all the WGS/WES data in GCAD. It is a functional equivalent pipeline jointly developed by GCAD/ADSP and CCDG/TOPMed. It outputs a CRAM (after recalibration and indel realignment) as well as a gVCF (generated using GATK haplotypecaller).
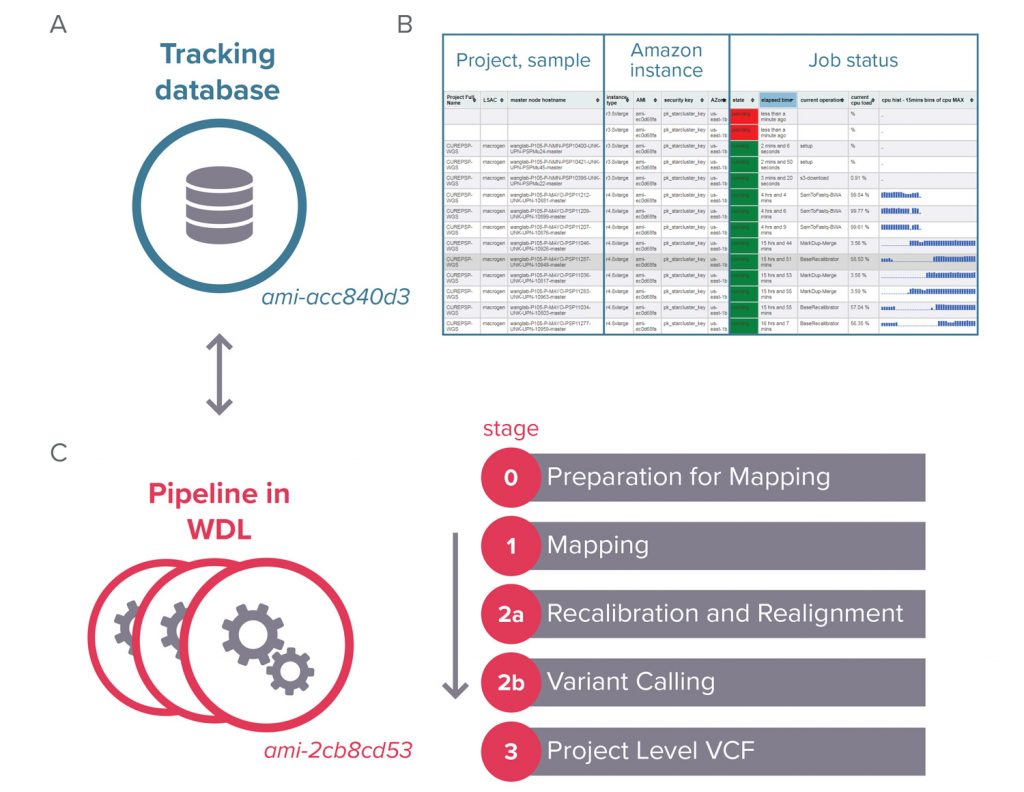
For more information go to VCPA page.
QC pipeline
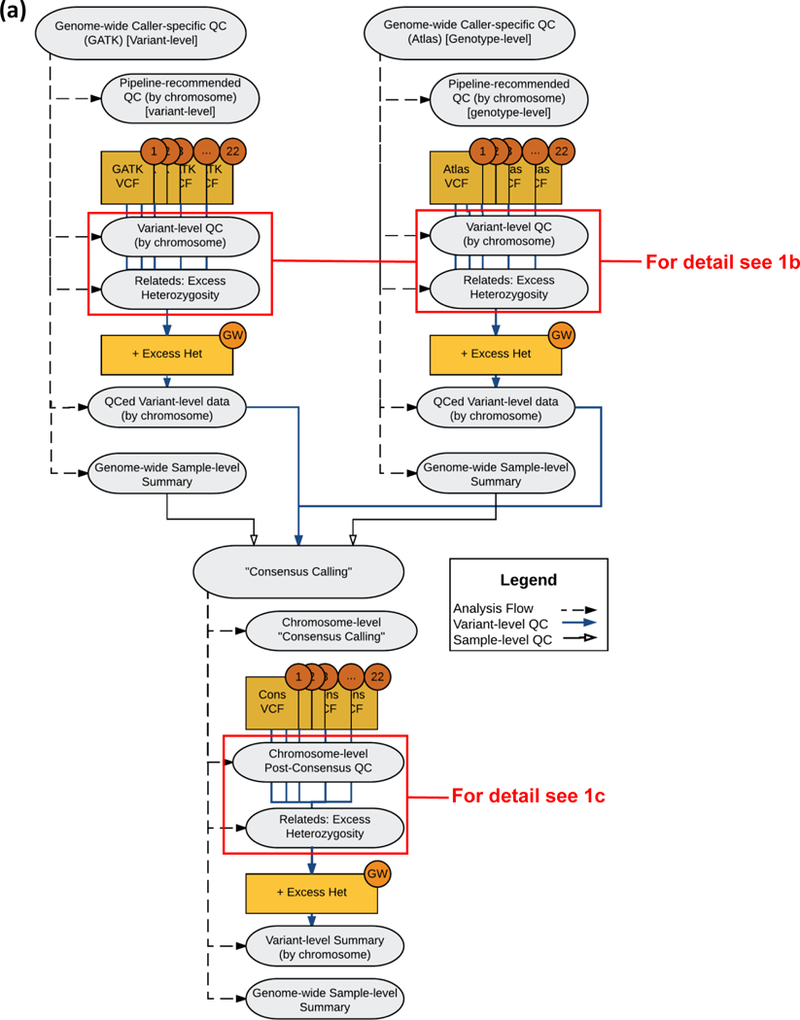
Project level VCF is QC-ed via a multi-stage process. 1) pre-QC quality checks are performed, including concordance with GWAS data, sample contamination, relatedness/duplication, and Mendelian inconsistency. 2) Individual genotypes, variants, and samples’ checks (e.g. average read depth, average genotype quality scores, and departure from Hardy-Weinberg Equilibrium) are done next. Variants are flagged when issues arise. 3) Finally, improvements are assessed based on quality with the exclusion of low-quality genotypes, variants, and samples as flagged in the second stage.
To learn more about QC Pipeline please read our publication.
Annotation pipeline
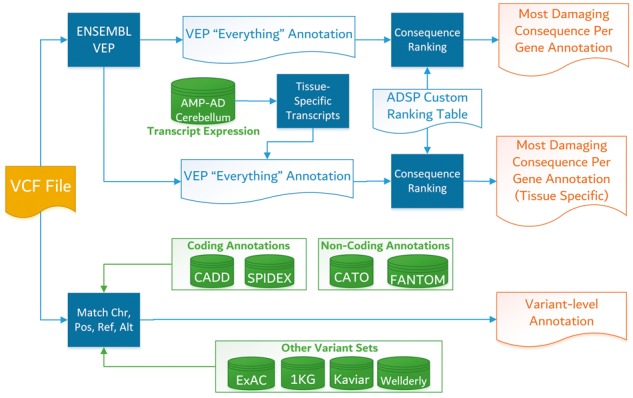
The pipeline generates variant-level assessments of functional impact on genes and genetic regulation. Our pipeline is based upon the Ensembl Variant Effect Predictor, which overlays exon, transcript, and regulatory element information from the Ensembl database to generate all possible consequences (missense, frameshift, splicing, etc) a variant may have. Variant consequences relative to Ensembl/GENCODE transcripts are assigned an impact category (high, moderate, low, etc), and multiple variant scoring approaches are incorporated (CADD, CATO, etc).
Learn more about GCAD annotation pipeline.